While one patient may walk through your door with a classic clinical picture, leading to an easy diagnosis, another may present with signs and symptoms that have you phoning a friend. The mystery cases presented here can help to hone your diagnostic skills and prepare for even the most surprising retinal conditions.
- Rebecca Hepp, Editor-in-Chief
MYSTERY CASE NO. 1
By Meera D. Sivalingam, MD; Taku Wakabayashi, MD, PhD; and Yoshihiro Yonekawa, MD
An 11-year-old boy with a history of microcephaly and developmental delay was referred for retinal screening. His VA was 20/40 in each eye. Fundus examination and widefield fluorescein angiography (FA) demonstrated avascular peripheral retina and geographic chorioretinal atrophy in the inferior midperiphery of each eye (Figure 1). OCT showed well-preserved inner and outer retina laminations in the central macula, but the outer retina became attenuated leading up to the inferior area of atrophy (Figure 2).
What do you think is the diagnosis? See the discussion below to find out.
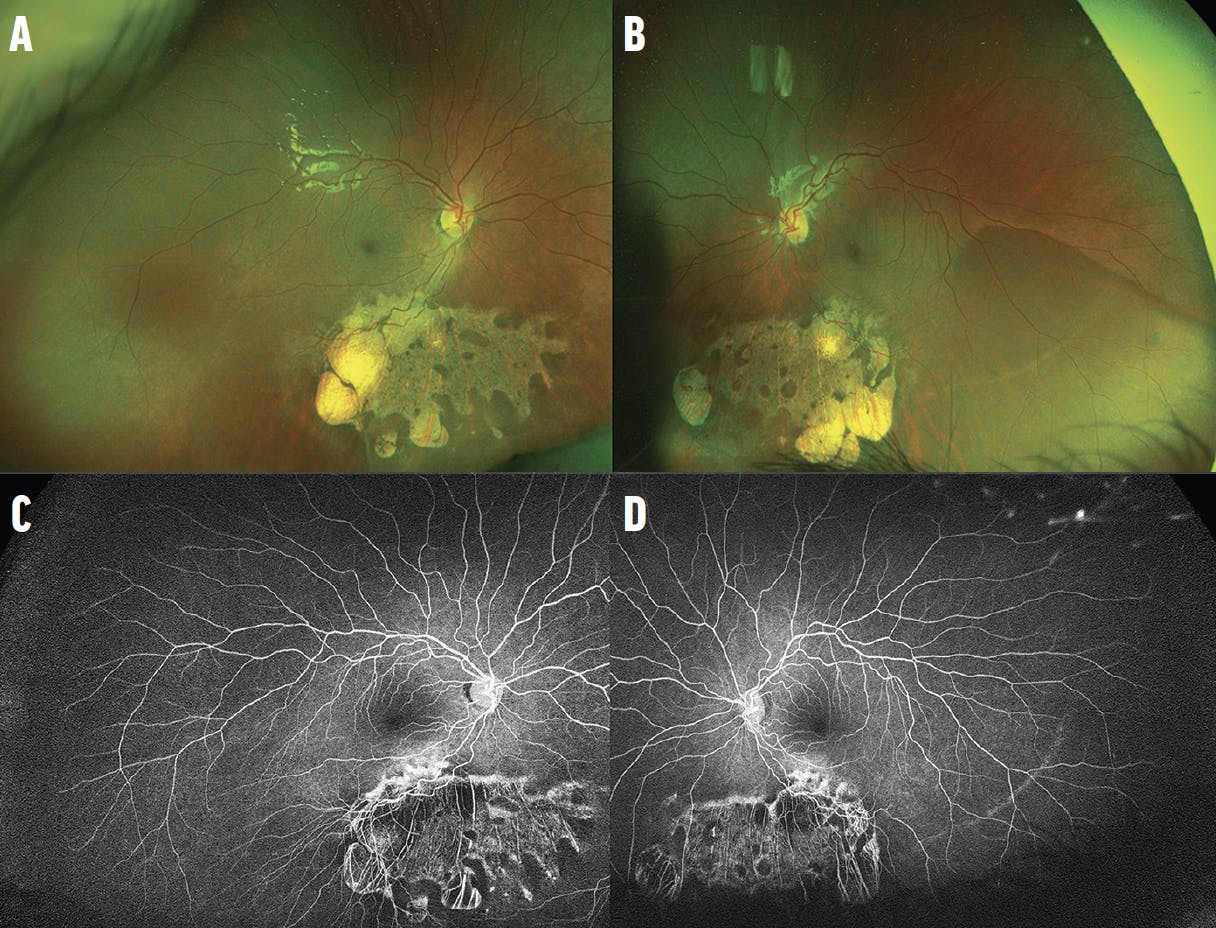
Figure 1. Widefield imaging of the right (A) and left (B) eyes shows peripheral avascular retina, retinal dragging, and midperipheral chorioretinal atrophy. Widefield FA of the right (C) and left (D) eyes highlights peripheral avascular retina and the chorioretinal atrophy. There is no neovascularization.

Figure 2. Spectral-domain OCT of the right (A) and left (B) eyes shows that overall microanatomy of the macula is intact, except for the start of outer retinal attenuation approaching the area of atrophy inferiorly.
MYSTERY CASE NO. 2
By Taku Wakabayashi, MD, PhD; Meera D. Sivalingam, MD; and Yoshihiro Yonekawa, MD
A 13-year-old boy presented with decreased vision in the left eye. VA was 20/20 OD and 20/200 OS. The fundus examination of the right eye was unremarkable, while the left eye showed an optic disc anomaly (Figure 3). FA showed hyperfluorescence around the disc and midperiphery corresponding to areas of chorioretinal atrophy. Retinal nonperfusion was also observed in the temporal periphery. OCT showed peripapillary serous subretinal fluid. Brain MRI and MRA did not show evidence of Moyamoya disease.

Figure 3. Fundus imaging shows an enlarged disc with radiating retinal vasculature (A). FA highlights the retinal vascular pattern, nonperfusion, and areas of chorioretinal atrophy (B). A magnified image helps depict the optic disc (C). Vertical OCT of the disc shows peripapillary subretinal fluid (D, yellow asterisks). Horizontal OCT shows a thin macula without foveal microanatomy (E).
What is causing this patient’s decreased vision? Find out in the discussion below.
MYSTERY CASE NO. 3
By Natasha Ferreira Santos da Cruz, MD; Carlos Ernesto Mendoza Santiesteban, MD; and Audina M. Berrocal, MD
An 8-year-old boy was referred to the office with problems with his night vision. The patient’s mother reported that the child prefers brighter rooms. The patient had a normal birth history and no past ocular or family history. On initial examination, the patient's BCVA was 20/25 OD and 20/30 OS. The anterior segment examination was normal in each eye. The fundus examination revealed diffuse and discrete whitish flecks with macular sparing in each eye (Figure 4). OCT imaging located the lesions in the zone of interaction between the photoreceptor inner and outer segment and the apical retinal pigment epithelium (RPE, Figure 5). Fundus autofluorescence imaging showed small flecks of hyperautofluorescence in each eye, which may correlate to the spots on the fundus photographs. Full-field electroretinogram (ERG) testing demonstrated diminished scotopic responses that markedly improved to normal after prolonged dark adaptation.

Figure 4. The patient's fundus photographs document diffuse, discrete, macula-sparing whitish flecks in the right (A) and left (B) eyes.

Figure 5. OCT imaging shows the lesions in the zone of interaction between the photoreceptor inner and outer segment and the apical RPE (white arrows) in the right (A) and left (B) eyes.
What is causing this child’s night vision problems? See the discussion below to find out.
WE ASKED, YOU ANSWERED
Retina specialists took to social media to sleuth out the answers to these mystery cases. How well did you do?



DISCUSSION
Case No. 1: KIF11 Vitreoretinopathy
Genetic testing in this patient revealed a heterozygous mutation in KIF11 (c.128_131dup), which was predicted to result in a frameshift and premature protein termination. The findings have been stable during subsequent follow-up visits.
Familial exudative vitreoretinopathy (FEVR) is a group of inherited retinal diseases characterized by abnormal retinal vascular development. Early stages are often asymptomatic and characterized by peripheral avascular retina and anomalous retinal vasculature. More advanced stages result in neovascularization, exudation, and tractional retinal detachment. FEVR is caused by genetic mutations in the Wnt signalizing pathway. The classic genes are NDP, FZD4, TSPAN12, and LRP5, but in recent years, researchers have identified additional genes implicated in FEVR-like syndromes.1,2
Clinicians are encouraged to identify the genetic etiology because there are different systemic consequences. For our patient described above, the KIF11 mutation causes a FEVR-like phenotype with microcephaly, developmental delay, and chorioretinal atrophy. It is a part of a syndrome called microcephaly with or without chorioretinopathy, lymphedema, or intellectual disability, or MCLID.1,2
Case No. 2: Morning Glory Disc Anomaly (MGDA)
Also known as morning glory syndrome, this congenital anomaly of the optic disc is characterized by an enlarged, funnel-shaped excavation of the optic disc (megalopapilla), glial tufts within the disc, and peripapillary chorioretinal pigmentary changes.3 An aberrant radial disposition of retinal vessels originating from the disc is also characteristic.
The diagnosis of MGDA is primarily made by the characteristic optic disc findings. Possible concomitant ophthalmic comorbidities include strabismus, retinal detachment (serous or rhegmatogenous), persistent fetal vasculature, cataract, aniridia, microphthalmos, peripheral retinal nonperfusion, and peripapillary choroidal neovascularization.4 MGDA is typically unilateral, but bilateral involvement is also reported. Visual prognosis is often limited with a VA ranging between 20/200 and counting fingers in the affected eye, although some eyes retain good vision.
It is important to rule out Moyamoya disease in patients with MGDA because this association is common and can be life threatening. Other systemic associations may include basal encephalocele, PHACES syndrome, Aicardi syndrome, Chiari malformation type I, neurofibromatosis type 2, and CHARGE syndrome.
Case No. 3: Biallelic RDH5 Mutation in Fundus Albipunctatus
This patient’s genetic testing was positive for two variants in the RDH5 gene, p.Arg54Ter and p.Arg191Gln, confirming the diagnosis of fundus albipunctatus. After 1 year of follow-up, the patient’s retinal findings have remained stable.
Fundus albipunctatus is a rare autosomal recessive form of congenital stationary night blindness that is characterized by night blindness from childhood, stationary or slow progression of rod abnormalities, and progressive cone abnormalities in older age. Patients usually complain of delayed dark adaptation after exposure to bright light with normal visual acuity and color perception.5-7
The disease is characterized by numerous small white dots from the midperipheral to peripheral retina, without vascular or optic nerve abnormalities. The deposits are localized between the outer limiting membrane and the outer aspect of the RPE on OCT.8,9 The shape and number of spots will vary with age and may even fade entirely.10 The presumed accumulation of cisretinol and cisretinyl esters in the RPE due to 11-cis-retinol dehydrogenase deficiency may be responsible for the formation of the white dots seen in RDH5 mutation-associated fundus albipunctatus.11
The differential diagnoses of flecked retinal disorders include retinitis punctata albescens, vitamin A deficiency, dominant radial drusen, benign familial fleck retina, fundus flavimaculatus, and fleck retina of Kandori; however, a much wider array of diseases correspond to the vague definition of fleck retina syndromes. The extension of flecks to the retinal periphery, lack of drusen on the macula and the nasal side of the optic disc, and the absence of high-density autofluorescent deposits exclude dominant radial drusen and fundus flavimaculatus.
Although benign retinal flecks show normal results in a full-field ERG, prolonged or overnight dark adaptation should be performed to differentiate fundus albipunctatus and retinitis punctata albescens. The ERG result normalizes or improves after prolonged dark adaptation for patients with fundus albipunctatus, while the result remains abnormal even with prolonged dark adaptation for patients with retinitis punctata albescens.12
The case presented here highlights the importance of considering fundus albipunctatus in the differential diagnosis of retinal flecks disease. For patients suspected to have this condition, appropriate genetic analysis and electrophysiological findings appear to be crucial elements of a proper diagnosis.
1. Hu H, Xiao X, Li S, Jia X, Guo X, Zhang Q. KIF11 mutations are common causes of autosomal dominant familial exudative vitreoretinopathy. Br J Ophthalmol. 2016:100(2):278-283.
2. Li JK, Fei P, Li Y, et al. Identification of novel KIF11 mutations in a patient with familial exudative vitreoretinopathy and phenotypic analysis. Sci Rep. 2016;6:26564.
3. Kindler P. Morning glory syndrome: unusual congenital optic disk anomaly. Am J Ophthalmol. 1970;69(3):376-384.
4. Haik BG, Greenstein SH, Smith ME, Abramson DH, Ellsworth RM. Retinal detachment in the morning glory anomaly. Ophthalmology. 1984;91(12):1638-1647.
5. Marmor MF. Fundus albipunctatus: a clinical study of the fundus lesions, the physiologic deficit, and the vitamin A metabolism. Doc Ophthalmol. 1977;43:277-302.
6. Schatz P, Preising M, Lorenz B, Sander B, Larsen M, Rosenberg T. Fundus albipunctatus associated with compound heterozygous mutations in RPE65. Ophthalmology. 2011;118(5):888-894.
7. Liu X, Liu L, Li H, Xu F, Jiang R, Sui R. RDH5 retinopathy (fundus albipunctatus) with preserved rod function. Retina. 2015;35(3):582-589.
8. Sergouniotis PI, Sohn EH, Li Z, et al. Phenotypic variability in RDH5 retinopathy (fundus albipunctatus). Ophthalmology. 2011;118(8):1661-1670.
9. Schatz P, Preising M, Lorenz B, et al. Lack of autofluorescence in fundus albipunctatus associated with mutations in RDH5. Retina. 2010;30(10):1704-1713.
10. Sekiya K, Nakazawa M, Ohguro H, Usui T, Tanimoto N, Abe H. Long-term fundus changes due to fundus albipunctatus associated with mutations in the RDH5 gene. Arch Ophthalmol. 2003;121(7):1057-1059.
11. Driessen CA, Winkens HJ, Hoffmann K, et al. Disruption of the 11-cis-retinol dehydrogenase gene leads to accumulation of cis-retinols and cis-retinyl esters. Mol Cell Biol. 2000;20(12):4275-4287.
12. Wang NK, Chuang LH, Lai CC, et al. Multimodal fundus imaging in fundus albipunctatus with RDH5 mutation: a newly identified compound heterozygous mutation and review of the literature. Doc Ophthalmol. 2012;125(1):51-62.



_1784132761.jpg?auto=compress,format&w=75)











